 TR
TR
 EN
EN
 DE
DE
 RU
RU
 AR
AR

"SMA VE Sağlıklı Bebek Arzusu: Ne zaman testler önerilmeli"
Spinal Musküler Atrofi (SMA), doğuştan bir genetik bozukluk nedeniyle ortaya çıkan ve omurilik ön boynuzda bulunan alfa motor nöronları tutan bir hastalıktır (1). Omurilik ön boynuzda bulunan alfa motor nöronlar hareketten sorumludur ve doğrudan vücudun iskelet kaslarını kontrol eder (Şekil 1).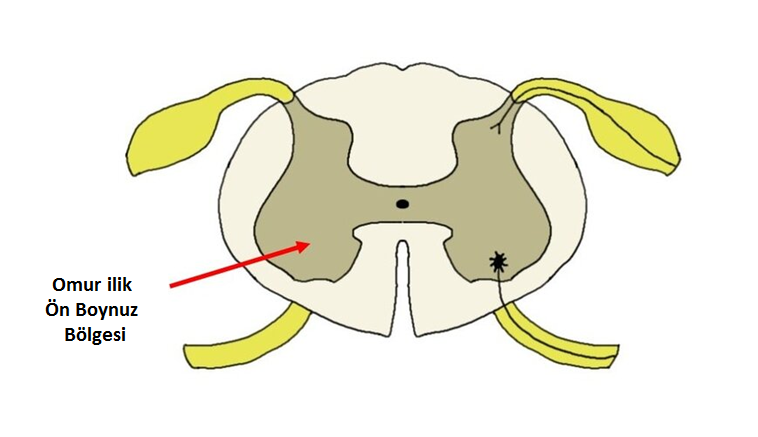
Şekil 1. Omurilik alfa motor nöronların bulunduğu ön boynuz bölgesi
SMA kalıtsal genetik bir hastalıktır. Genetik hastalıklar içinde tek gen hastalıkları olarak adlandırılan grupta yer alır. Yaklaşık hepimiz 20.000 gene sahibiz ve bu genlerden biri annemizden, biri de babamızdan olmak üzere, her bir genden ikişer adet bulunmaktadır. Tek gen hastalıkları dediğimizde, bu genlerden birinin fonksiyon görmemesi sonucu meydana gelen hastalıkların aklımıza gelmesi gerekiyor. SMA da bu hastalıklardan biridir. Tek gen hastalıkları da kendi içinde sınıflara ayrılmaktadır. Bu sınıflar içinde ise SMA otozomal resesif (çekinik) tek gen hastalıkları içine girmektedir. Otozomal resesif hastalıklarda hastalığın ortaya çıkması için her iki gende de mutasyon sonucu fonksiyon bozukluğu olması gerekmektedir. Tek bir gende mutasyon olması hastalık belirtilerini ortaya çıkarmaz. Tek bir gende mutasyonu olan kişilere taşıyıcı denir. Otozomal resesif tek gen hastalıklar çoğu zaman iki taşıyıcı çiftin çocuklarında görülmektedir. Bu çiftlerin çocuklarının SMA olma olasılığı %25, taşıyıcı olma olasılığı %50 ve her iki geninde de mutasyon olmama olasılığı yine %25’tir (Şekil 2). SMA’da da genellikle böyle olmaktadır. SMA, 5. kromozom üzerindeki SMN1 adı verilen bir genin genetik kusurlarından kaynaklanır (2). Her 40-60 kişiden biri, SMA'ya neden olan gen varyasyonunun taşıyıcısıdır.
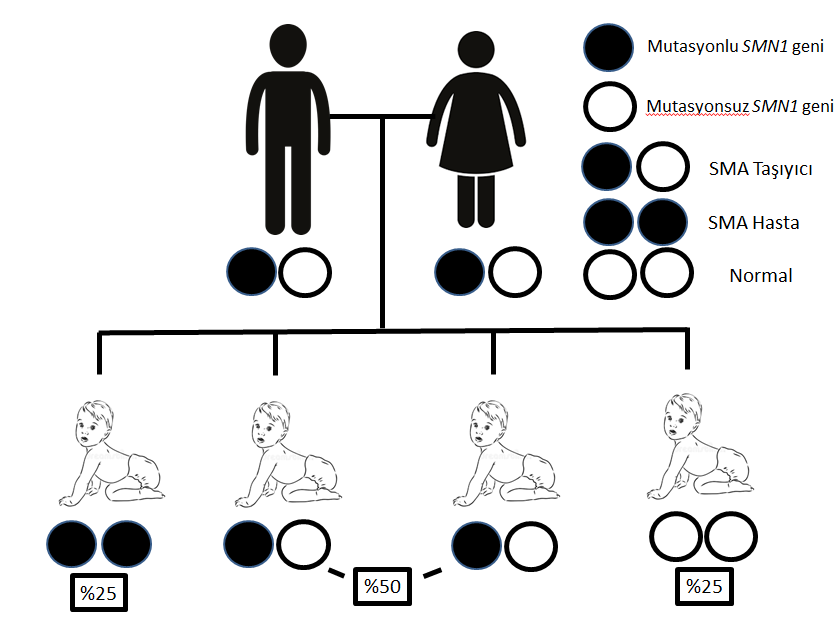
Şekil 2. SMA’da otozomal resesif kalıtım şekli
Bu gen, SMN denilen proteinin sentezini yapar. Yeterli SMN proteini olmadığı takdirde, omurilik alfa motor nöronları küçülmeye ve ölmeye başlar. Bunun sonucunda beyin özellikle bizim isteğimizle hareketini sağladığımız kol, bacak, baş ve boyun kaslarını kontrol edemez. Kullanılamayan kaslar zamanla zayıflamaya başlar. Bu durum yürüme, emekleme, baş ve boyun kontrolü, yutma ve nefes alma gibi hareketlerin yapılamaması ile sonuçlanır.
SMA hastalığının görülme oranı 1/10000’dir. İkinci en sık görülen otozomal resesif hastalıktır (1). SMA hastalığının 5 tane alt tipi vardır. Bu tipler 0 ile 4 arasında numaralandırılır. Tip 0, en ağır klinik durumdan, Tip 4 en hafif klinik duruma doğru sınıflanırlar. Her bir tipin görülme sıklığı farklıdır. En sık Tip 1 SMA görülmektedir (Tablo 1).
Tablo 1. SMA sınıflaması
|
SMA Tipi |
Görülme % |
Başlangıç |
Yaşam |
Klinik |
|
0 |
<1 |
Doğumda |
1-2 hafta |
Baş tutma ve oturma yok |
|
1 |
45 |
0-6 ay |
1-2 yaş |
Baş tutma var, oturma yok |
|
2 |
20 |
6-18 ay |
25 yaş |
Oturma var, yürüme ve desteksiz solunum yok |
|
3 |
30 |
18 ay- 30 yaş |
Normal |
Normal klinik |
|
4 |
5 |
30 yaş < |
Normal |
Normal klinik |
Genlerin Yapısı
Yukarıda da belirtildiği gibi SMA hastalığı SMN1 genindeki mutasyonlar sonucu görülür. SMN1 geninin sentezlediği SMN proteininin olmaması nedeniyle klinik bulgular ortaya çıkar. SMN proteinini sentezleyen tek gen vücudumuzda SMN1 geni değildir. SMN1 geni yapısına %99 benzerlik gösteren SMN2 geni de SMN proteini sentezlemektedir. Yalnız SMN2 geni çok az yapısal olarak kaliteli SMN proteini sentezi yapabilir. Bu da toplam SMN proteininin yaklaşık %10’nunu oluşturur (Şekil 3).
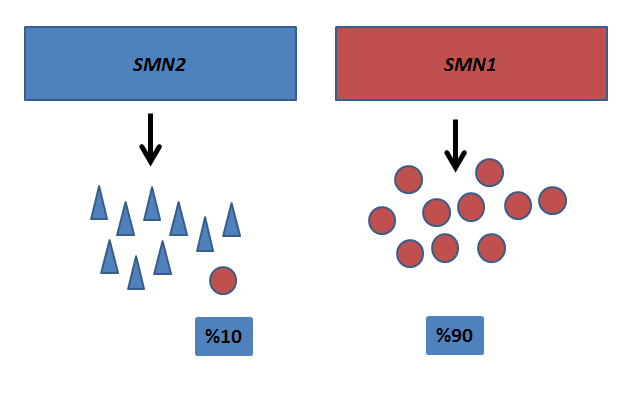
Şekil 3. SMN1 ve SMN2 genleri ve sentezledikleri SMN proteini oranları
Normalde her genimizden biri anneden, biri de babadan gelmek üzere iki adet bulunmaktadır. Bu kural SMN1 ve SMN2 genleri için geçerli değildir. SMN2 gen sayısı arttıkça sentezlenen SMN proteini oranı artacağı için SMA’ da klinik daha hafif olmaktadır. En sık görülen Tip 1 SMA’ da en fazla 1 veya 2 kopya SMN2 geni görülürken, Tip 2 ve yukarısında en az 3 kopya SMN2 geni bulunur. SMN1 geni ise çoğu zaman iki kopya bulunurken, bazı insanlarda 3 kopya olarak bulunabilir.
SMN1 geni mutasyonları
Genleri bir kitap gibi düşünürsek, genlerin kendi içlerinde protein kodlayan kısımları kitap bölümleri gibi ayrılmaktadır. Her genin farklı sayıda bölümleri vardır. SMN1 ve SMN2 genleri 8 bölümden oluşmaktadır ve bu bölümlerin sadece 7’si protein kodlaması yapmaktadır. Bu bölümlerin her birine ekzon denir. SMA hastaların yaklaşık %95’inde 7. bölümün (ekzonun) tamamen kaybının (delesyon) söz konusu olan mutasyon görülür. Hastaların sadece %5’inde kitaptaki bir harf değişikliğine benzetebileceğimiz nokta mutasyonları gözükmektedir. Nasıl bir mutasyon görülürse görülsün, SMN1 geninin fonksiyonu bozulur ve SMN proteini sentezlenemez hale gelir (Şekil 4).
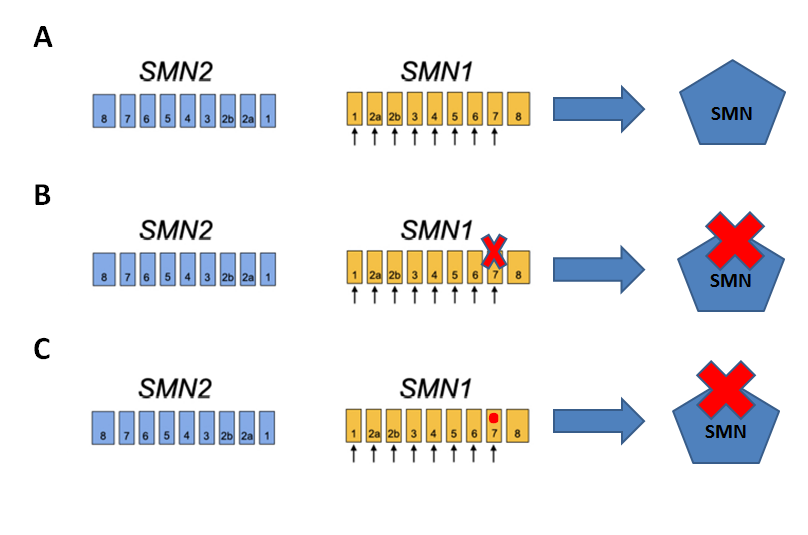
Şekil 4. SMN1 ve SMN2 geni yapısı; A) Normal gen yapısı, B) En sık görülen ekzon 7 mutasyonu, C) Nokta mutasyonu
SMA’da kullanılan genetik testler
SMA hastalarında yukarıda da belirtildiği gibi %95 hastada ekzon 7 kaybı görüldüğü için bu kaybı göstermeye yönelik genetik testler kullanılır. Bu yöntemler kolay ve hızlı sonuç veren yöntemlerdir, fakat %5 hastada görülen nokta mutasyonlarını göstermez. Hastanın kliniği SMA ile uyumlu ise delesyonu gösterilemese bile mutlaka nokta mutasyonları açısından SMN1 geninin tamamı taranmalıdır.
Yenidoğan Taramaları
Ülkemizde SMA için rutin bir Yenidoğan (YD) taraması yoktur. Yalnız sayılı ülkeler SMA’ yı yenidoğan taramasına almışlardır. Bir hastalığın yenidoğan taramasına girmesi için; önemli bir sağlık sorunu olması, YD döneminde tanı olasılığı olması, uygun uygulanabilir bir testinin olması ve erken tedavinin etkili olduğu bir hastalık olması gerekmektedir. SMA hastalığı bu özelliklerin hepsini kapsamaktadır. Özellikle en sık görülen SMA tip 1 hastaları ilk 3 ayda alfa motor nöronlar hızla yıkılmaya başlamaktadır ve ne kadar erken tedavi başlanırsa tedaviye o kadar iyi cevap vermektedirler. İlk 3 ay sonrasında verilen tedaviler ne yazık ki hangi tedavi olursa olsun çok etkili olamamaktadır. SMA’ da biyokimyasal belirteç olmadığı için tarama genetik testlerle yapılmaktadır. Tarama yapılan ülkelerde daha çok en sık görülen delesyonu mutasyonu taranmaktadır. Bu daha az görülen nokta mutasyonlarının tanı almaması ve hastaların gözden kaçmasına neden olmaktadır. Günümüzde tüm genom dizileme (WGS) denilen yöntem ile tüm DNA’mız ilk harfinden son harfine kadar okunmaktadır. Bu yöntem ile hem delesyonlar hem de nokta mutasyonları aynı anda gösterilmektedir. Günümüzde halen çok pahalı olan bu yöntemin rutin taramalarda kullanılması şu an için zor gözükmektedir. İleriki yıllarda sadece SMA taramasında değil birçok hastalığın taramasında kullanılacağına inanılmaktadır.
Taşıyıcılık Testi
Ülkemiz gibi akraba evliliklerinin yüksek görüldüğü ülkelerde özellikle akraba evliliği yapan çiftlere mutlaka önerilmesi gerekmektedir. Sadece SMA değil, tüm otozomal resesif geçişli tek gen hastalıkları akraba evliliklerinde yüksek oranda görülmektedir. Günümüzde en sık taşıyıcılık testi yapılma endikasyonu ise aile SMA’ lı bir olgu görülmesi sonucu olmaktadır. Ailenizde SMA olgusu var ise önce siz, taşıyıcı çıkarsanız eşiniz mutlaka SMA için taşıyıcılık testi yaptırma zorunluluğu vardır.
Taşıyıcı olanlar ne yapmalı?
Eşiniz ve siz SMA için taşıyıcı iseniz yapabileceğiniz başlıca 2 şey bulunmaktadır. Bunlardan birincisi normal gebeliği takiben anne karnında olan bebeğe yönelik test (prenatal tanı) yapılmasıdır. Bu da gebeliğin 11-13 haftasında koryonik villus biyopsisi (CVS) veya 16-18 haftaları arası amniyosentez şeklinde yapılabilir. Her iki test de riskli girişimlerdir, ufak da olsa düşük, enfeksiyon ve kanama gibi riskleri bulunmaktadır. Bu testler sonucunda bebeğe ait DNA’ dan yapılan genetik analiz ile bebeğin SMA hastası olup olmadığı %99.7’ ye yakın doğrulukla belirlenmektedir. Bu testin en önemli dezavantajı ise anne karnında SMA tanısı konan bebeğe bu dönemde bir tedavi olmamasıdır. Ancak günümüzde SMA’ lı bebeğe doğar doğmaz başlanacak tedaviler ile yüz güldürücü sonuçlar alınabileceği akılda tutulmalıdır.
İkinci yöntem ise tüp bebek eşliğinde yapılan yöntemdir. Beşinci güne gelen embriyolardan parça alınarak, hasta, taşıyıcı ve normal embriyolar tespit edilir. Taşıyıcı ve/veya normal embriyoların transferi yapılarak sağlıklı çocukların doğması sağlanmaktadır. Bu yöntem de tanı açısından son derece güvenilir bir genetik yöntem olsa da günümüzde halen prenatal tanı ile her zaman doğrulama yapılması önerilmektedir.
SMA, bir asırdan uzun zamandır tanımlanmış bir hastalık olmakla birlikte günümüzde genetik tanı yöntemlerinin hızla gelişmesi, yüz güldürücü tedavilerin hızla kullanıma geçmesi ve tarama testlerinin devreye girmesi ile kontrolü mümkün bir hastalık olma yolunda hızla ilerleyeceği düşünülmektedir.
- Gönderiyi Paylaş







